Solid Abdominal Masses in Children
Erik Beek, Martine van Grotel, Bart de Keizer, Annemieke Littooij and Rutger Jan Nievelstein
Department of Radiology, Solid tumors and Nuclear Medicine of the University Medical Center Utrecht and Princess Maxima Center for Pediatric Oncology
Publicationdate
Malignant abdominal tumors in children are rare and usually present as solid masses. Treatment is done in specialized centers, but the initial diagnosis is usually made in the hospital where the child first presents.
In this article we will provide tools to make the initial imaging diagnosis of the most common malignant abdominal tumors as accurate as possible. This will guide the next imaging procedures
Some benign tumors which can easily be mistaken for a malignancy are included.
Cystic abdominal masses in children are discussed here.
Introduction

The most common cancers overall in children are leukemia (28%), brain and spinal tumors (26%).
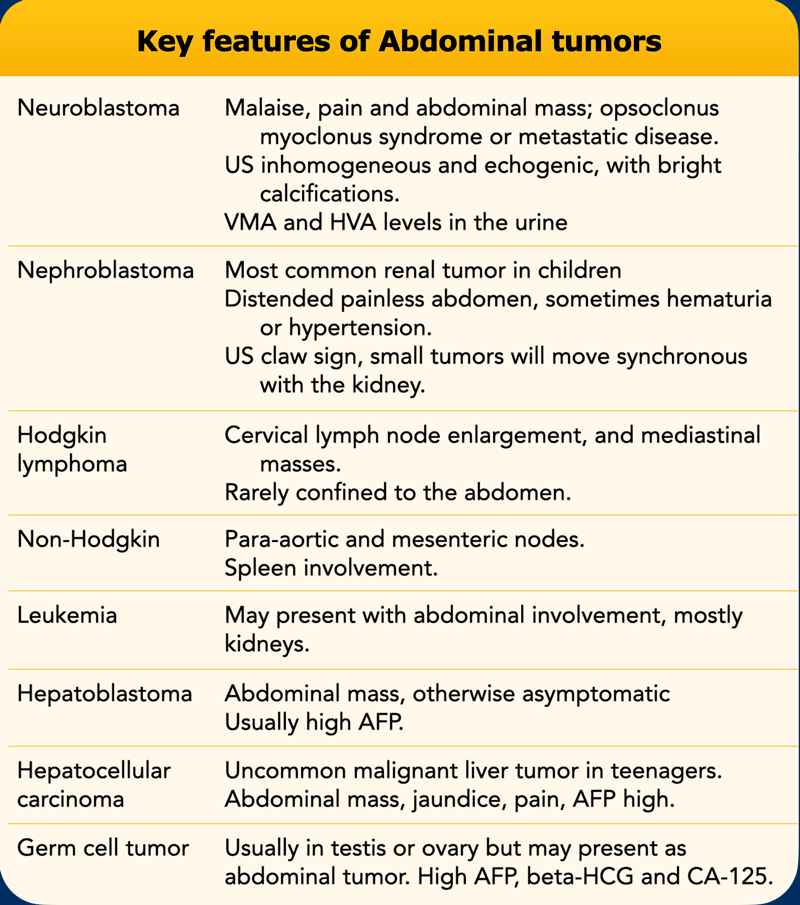
They are followed by tumors that mainly present as an abdominal tumor: Neuroblastoma (8%), Nephroblastoma (5%) and Lymphoma (8%) (ref).
Pediatric abdominal tumors are often very large at initial presentation, because most children come to attention because someone noted severe abdominal distention.
It may seem a contradiction, but in very large tumors, it is usually more difficult to ascertain the organ of origin.
The most common intra-abdominal tumors in children are:
- Neuroblastoma: 30% of all cases
- Nephroblastoma or Wilms' tumor: 25%
- Lymphomas:15%
- Germ cell tumors: 9%
- Hepatoblastoma: 9%
- Rhabdomyosarcoma: 4%
- Hepatocellular carcinoma: 1,5%
The key features of these abdominal tumors are presented in the table.
Ultrasound is the first imaging modality to be used.
It can easily confirm that there is a mass and can often define the site of origin.
Search for synchronous movement with respiration of the tumor with its organ of origin.
This can be seen in smaller hepatic and renal tumors.
In large tumors the surrounding organs are compressed and will show no movement.
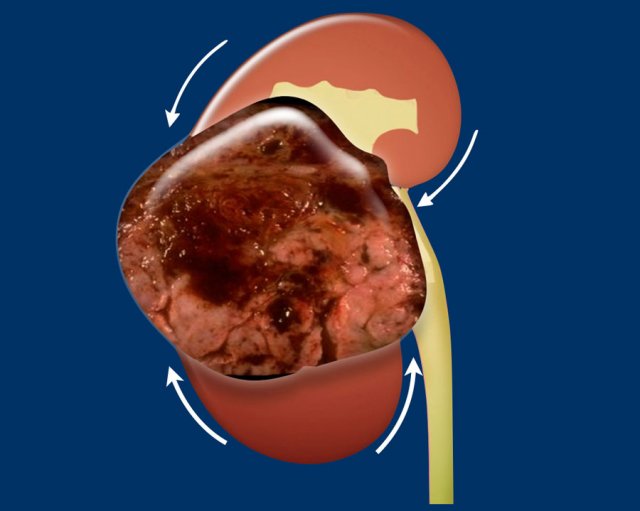 Claw sign in renal tumor
Claw sign in renal tumor
In renal tumors the “claw sign” is often present.
This is seen when a part of the kidney is draped around the tumor like a claw (figure).
Before we discuss the different abdominal tumors, it is good to realize that the definitive diagnosis is usually made by pathologic tissue examination.
Most tumors are biopsied before treatment.
However renal tumors in children between six months and nine years are not biopsied because the likelihood of it being a nephroblastoma is so high that the risk of a wrong diagnosis outweighs the risk of tumor spill during a biopsy, especially in diffuse anaplastic nephroblastoma.
Neuroblastoma

Neuroblastomas are embryonic tumors originating from the sympatho-adrenal lineage of the neural crest.
About half of the tumors arise from the adrenal glands.
Other sites of origin are the thoracic and lumbal paravertebral sympathic chain.
A minority develops in the neck.
The clinical presentation is variable. Common complaints are pain and an abdominal mass. Neurologic signs due to intraspinal extension can occur. A typical manifestation is “raccoon eyes”, which is periorbital ecchymosis due to metastatic infiltration of the orbital area.
A minority of the patients present with opsoclonus myoclonus syndrome, a neurological disorder characterized by rapid, multi-directional eye movements (opsoclonus), quick, involuntary muscle jerks (myoclonus), uncoordinated movement (ataxia), irritability, and sleep disturbance.
The
prognosis depends on the stage of the tumor.
For a low grade tumor the 5-year
survival is > 90%.
For high risk tumors (stage 4 and tumors with MYCN
amplification) it is around 50%.
The staging is rather complicated, see reference staging neuroblastoma.
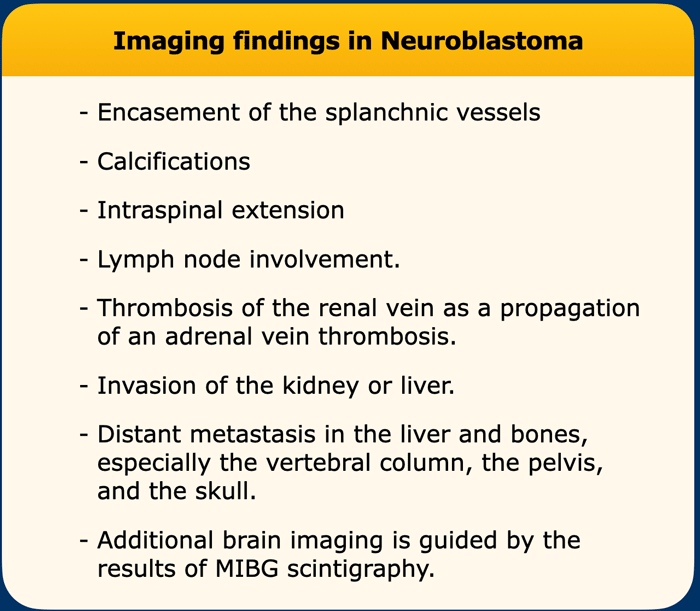
Imaging findings
The imaging findings are listed in the table.
Ultrasound
On Ultrasound the tumor is generally echogenic and inhomogeneous with bright calcifications.
A feature of neuroblastoma is the tendency of vascular encasement and they have a tendency to grow between the vertebral column and the aorta which is lifted ventrally.
Other tumors that show vascular encasement are lymphoma and rhabdomyosarcoma.
Intraspinal spread is common but difficult to assess with ultrasound.
If lymphatic spread has occurred many round tumorous lymph nodes can be seen. The left supraclavicular lymph node (Virchow’s node) is often involved in large left adrenal tumors.
Video
The video shows a neuroblastoma in a one year old boy who presented with vomiting. The tumor encircles the aorta which is lifted from the vertebral column. Small calcifications are present.
Continue with the MRI of this patient.
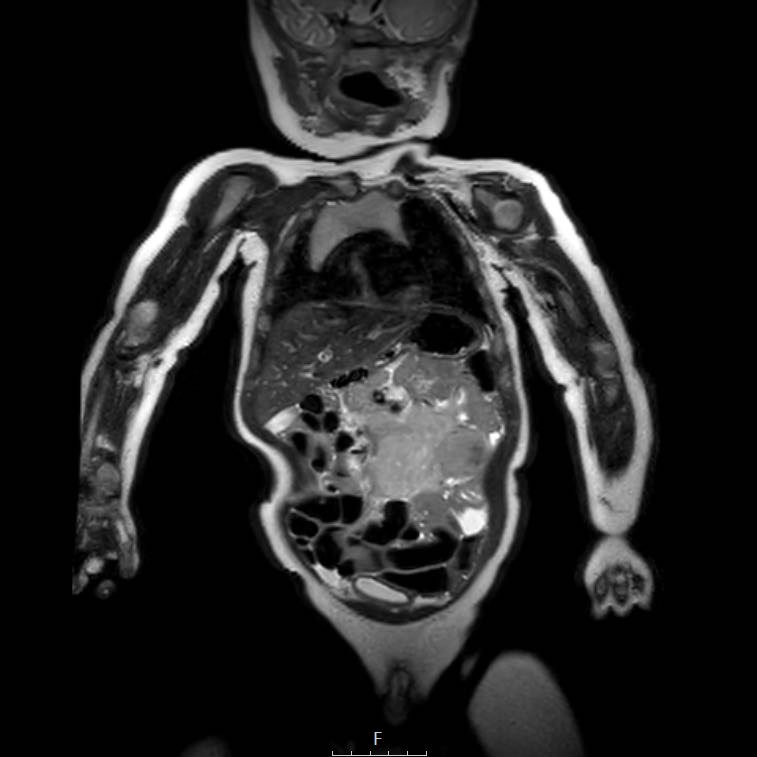
MRI
MRI is done for more detailed imaging of the tumor.
MRI examination:
- T2 weighted 3D sequence
- Fat suppressed T1 before and after Gadolinium injection
- Diffusion weighted imaging
Scroll through the coronal T2 weighted series.
Study the images and then continue reading.
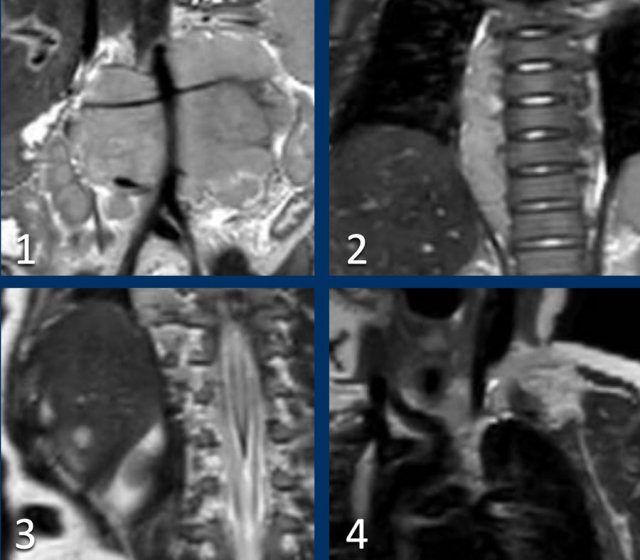
The findings are:
- Mass with encasement of the aorta and splanchnic vessels.
- Extension along the thoracic vertebral columns but no intraspinal invasion.
- Small liver metastases.
- Left supraclavicular mass.
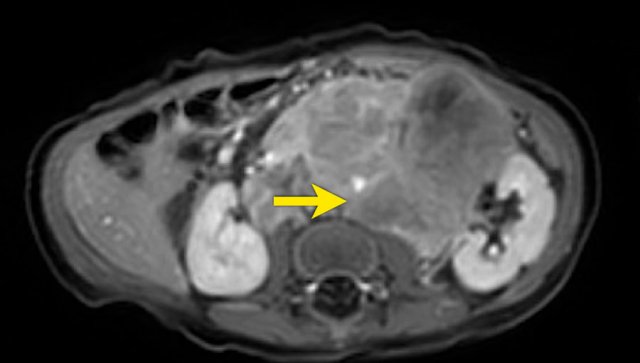
Same patient.
This is an axial gadolinium enhanced T1W-image with fat suppression.
It shows the encasement of the vessels.
Notice the tumor extension posterior to the aorta, which is displaced away from the vertebral column (arrow).
Ultrasound of a fifteen-month-old boy, who was first suspected of having a tumor in the left kidney.
Ultrasound shows a mass adjacent to the medial upper pole of the left kidney. It seems to be separate from the kidney. The mass is very inhomogeneous and has multiple calcifications.
These findings are more compatible with a neuroblastoma than a nephroblastoma.
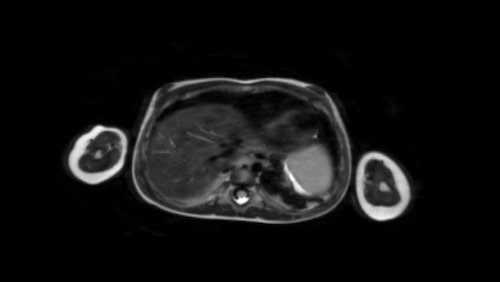
The extent of the tumor is well appreciated on a 3D axial T2 weighted TSE series.
The left kidney is compressed and displaced caudally.
Para-aortal lymph node metastases are present (small yellow arrow).
The origins of the celiac trunc and superior mesenteric artery are encased by tumor (arrowhead).
The inferior caval vein is lifted anteriorly (green arrow).
Bilateral dorsal atelectasis is often seen on the MRI, because the examination is done under anesthesia.
Biopsy
Percutaneous biopsy can be difficult for two reasons:
- These tumors contain much necrotic tissue resulting in only mucus-like material and not a pathology diagnosis
- These tumors can bleed heavily during biopsy. An open biopsy can be necessary, but that too can be accompanied by heavy bleeding.
Images
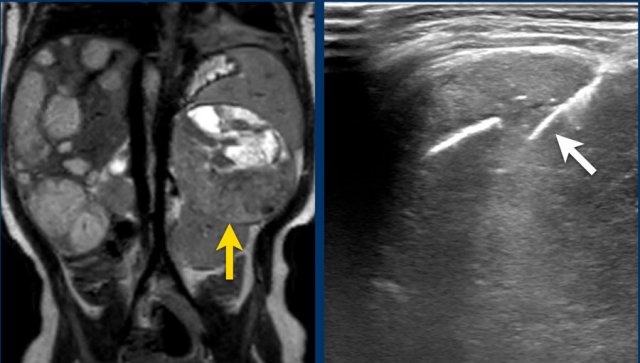
The MRI is of a nine-months-old girl with a tumor in the left abdomen. The MRI shows a tumor of the left adrenal gland, partially solid, partially cystic. Multiple liver metastases are present.
The tumor was biopsied. There was constant blood loss through the guiding needle. At the end of the procedure two gelatin foam plugs were placed (echogenic stripes (arrows).
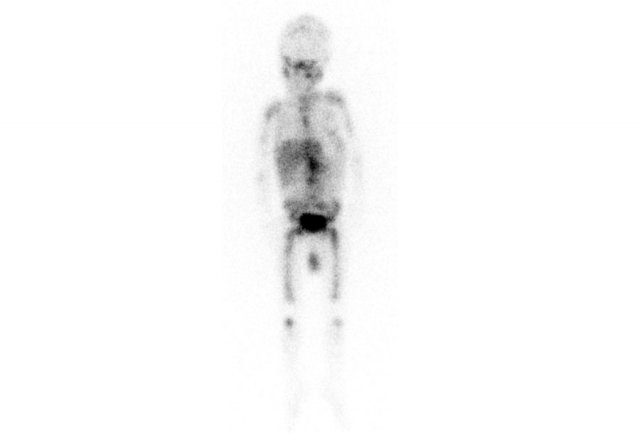
MIBG
All the activity indicates bone metastases.
In metastasized neuroblastoma follow-up imaging can be quite confusing, as bone metastases may become more apparent when they are successfully treated.
Renal tumors
Renal tumors in children will be discussed in more detail in a separate article.
Here we only show some common findings.
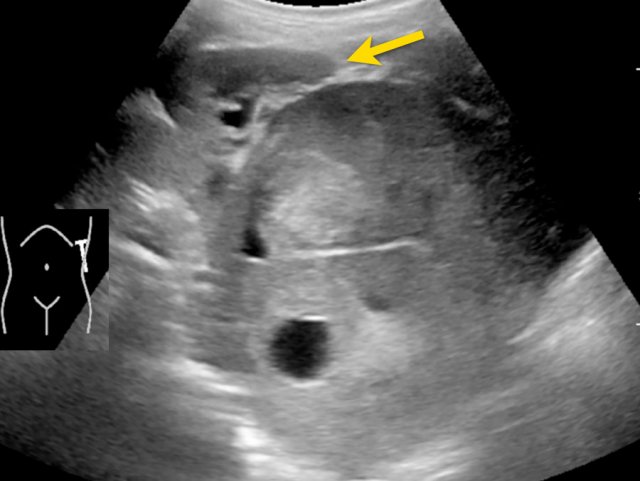
Nephroblastomas
More than 90% of renal tumors in children are nephroblastomas - also called Wilms tumor.
The peak age is 2 - 3 years.
Frequently these tumors present with a distended painless abdomen and they are often very large at presentation. Sometimes they present with hematuria, abdominal pain or hypertension.
Smaller tumors are found during sonographic screening in children with syndromes which predispose to nephroblastoma, like Beckwith-Wiedemann syndrome and Denys-Drash syndrome (see the webpage on renal tumors in children).
Bilateral nephroblastomas are often syndrome related.
The lungs are the most frequent site of metastases. Liver and bone metastases are rare.
Image
Nephroblastoma of the left kidney in a three-year-old boy. The remnant of the kidney is draped over the tumor (“claw sign” arrow). The tumor is rather homogeneous with some cystic areas.
Continue with the MRI.
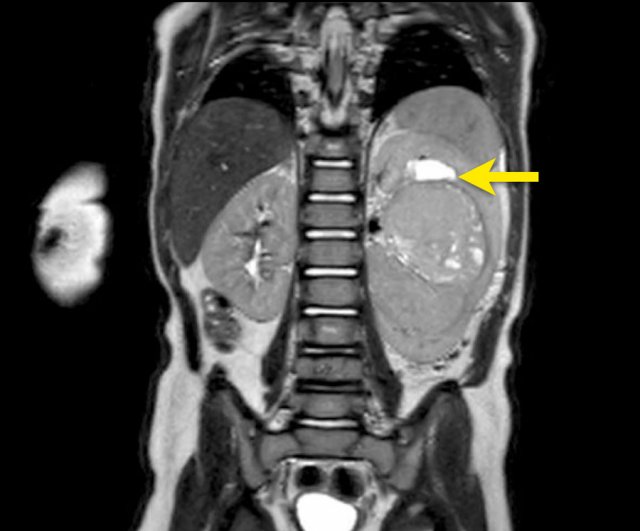
Image
Nephroblastoma of the left kidney in a three-year-old boy. A bilobar tumor is present in the interpolar region. There is a dilated calyx in the upper pole (arrow).
Treatment
In European countries the patient first receives chemotherapy, after which the kidney is resected, followed by post-operative chemotherapy (International Society of Pediatric Oncology, SIOP approach).
In the US, the kidney is primarily resected, followed by chemotherapy (Children’s Oncology Group, COG approach).
The results of both treatment regimens are about the same.
The prognosis is excellent with a 5-year survival of more than 90%.
Bilateral disease has a less favorable prognosis.
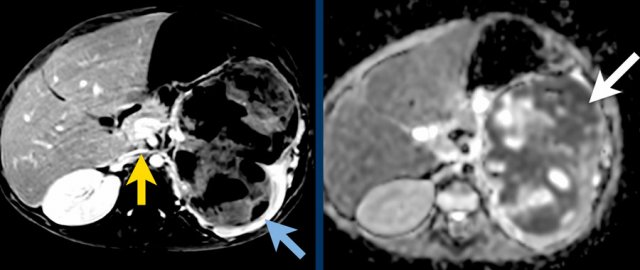 A. The tumor enhances less than the peripheral remnant of normal renal tissue (blue arrow). The left renal vein is open (yellow arrow). Solid parts of the tumor show diffusion restriction (white arrow)
A. The tumor enhances less than the peripheral remnant of normal renal tissue (blue arrow). The left renal vein is open (yellow arrow). Solid parts of the tumor show diffusion restriction (white arrow)
Ultrasound
The initial imaging is usually done by ultrasound. Smaller tumors will be seen to move synchronous with the kidney. Large tumors will not move.
As mentioned before, it is often possible to detect a remnant of the kidney draped around the tumor, the claw sign. The remnant can have a dilated calyx due to obstruction of the pelvis.
Small tumors are usually homogenic and echogenic.
Larger tumors are more inhomogeneous with cystic or necrotic parts and hemorrhage.
10% of the nephroblastomas have fine calcifications.
 Left sided nephroblastoma in a two-year-old girl. Note the para-aortal lymph node metastasis (arrow).
Left sided nephroblastoma in a two-year-old girl. Note the para-aortal lymph node metastasis (arrow).
Once you are sure of the renal origin of the tumor, study the other kidney for tumor or nephroblastomatosis (see below).
Search for enlarged para-aortal lymph nodes.
Search with color Doppler for patency of the renal vein and of the inferior caval vein, as a nephroblastoma tends to grow into the renal vein and inferior caval vein.
Also image the liver for metastases, although these are rare in nephroblastoma.
The finding of a liver metastasis should urge you to look for an alternative diagnosis like a rhabdoid tumor.
A CT chest is performed for the assessment of pulmonary metastases.
MRI
The next imaging step is a MRI of the abdomen.
Nephroblastomas are mostly inhomogeneous, with decreased signal intensity on T1 and increased signal intensity on T2. Necrotic cystic parts are often present.
Gadolinium enhancement is inhomogeneous and less than the enhancement of normal renal parenchyma.
Solid parts of the tumor will show restricted diffusion. Hemorrhage is often present. Hemorrhagic areas will also show restricted diffusion, so look on the T1-images for signs of bleeding.
Sometimes a disruption of the tumor capsule is seen. Intraperitoneal rupture is a more severe complication than retroperitoneal rupture.
MRI can nicely demonstrate tumor thrombus in the renal vein and inferior caval vein, and lymph node enlargement. It allows accurate and repeatable measurements of the tumor on the initial and follow-up examinations.
Example 1
A three-year-old girl with a tumor of the left kidney and a large tumor thrombus in the renal vein and inferior caval vein. Liver metastasis are present, (unusual finding) and a lung metastasis is seen.
Video of the same patient.
Notice the tumor thrombus in the left renal vein extending into the inferior caval vein.
Example 2
Video of a three-year-old girl with a large tumor in the right flank.
Ultrasound with a high frequency transducer shows that the tumor originates from the right kidney.
The remnant of the collecting system is dilated.
Same patient.
A tumor thrombus is present in the inferior caval vein.
Same patient.
An axial T2 weighted MR series shows the tumor thrombus extending towards the level of the hepatic veins. Note the dilated remains of the collecting system
Example 3
Video of a two-year-old boy with hemihypertrophy.
A screening ultrasound showed a homogeneous tumor in the upper pole of the left kidney.
Tumors discovered during screening examinations are usually much smaller.

Classification of Nephroblastoma
The classification of nephroblastomas is done after resection of the kidney.

Other Renal tumors
More renal tumors will be discussed in a separate article.
Liver tumors
Hemangioendothelioma
Hemangioendothelioma of the liver is also known as infantile hemangioendothelioma or infantile hepatic hemangioma. It is a highly vascular tumor. These tumors can be solitary, multiple or diffuse.
Most are discovered as an abdominal mass in the first six months of life. They can lead to congestive heart failure and rarely to Kasabach-Merritt syndrome, a rare disease, in which the vascular tumor leads to decreased platelet counts and bleeding disorder.
AFP levels are mostly normal.
Ultrasound
On ultrasound a well-perfused tumor is seen. It can be hypoechoic or of mixed echogenicity. Unlike adult hepatic hemangiomas they are not echogenic. Calcifications are common.
Large arteries and veins are seen and the aorta may be wider than normal due to the large demand of the tumor and tapers distal to the celiac axis.
Video
A three-month-old girl with a tumor in the left abdomen.
A tumor is visible with a stalk to the left liver lobe, in which large vessels are present. The lesion has some internal calcifications.
CT
On unenhanced CT calcifications are present in approximately half of the patients. After intravenous contrast the tumor shows peripheral enhancement with gradual filling-in. In larger tumors the center may not enhance at all.
MRI
On MRI a hemangioendothelioma has generally low signal intensity on T1 and high signal intensity on T2. After contrast the same filling-in is seen as on CT.
Most tumors will show spontaneous involution, and the prognosis is good.
Images
Scroll through the images. It is the same tumor as on the ultrasound.
Mesenchymal hamartoma
Mesenchymal hamartomas are usually multicystic liver lesions, although they can rarely be solid. They are often large at presentation. Serum AFP levels are normal.
Ultrasound will show a multicystic lesion. MRI will demonstrate this as well. After Gadolinium some stromal enhancement can be seen.
Example 1
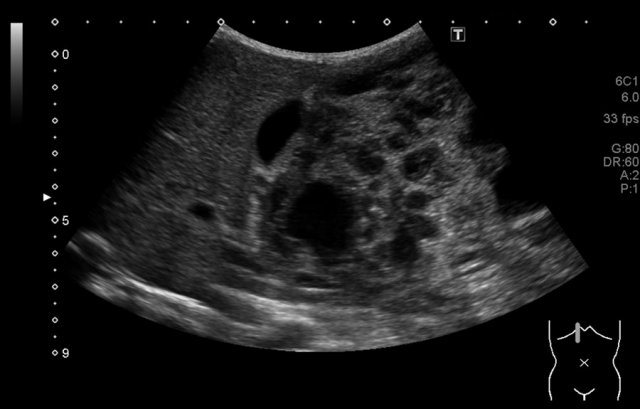
The image is of a two-year-old boy, who presented with a painless swelling of the abdomen. Ultrasound shows a large multicystic mass at the caudal side of the liver.
Continue with the MRI.
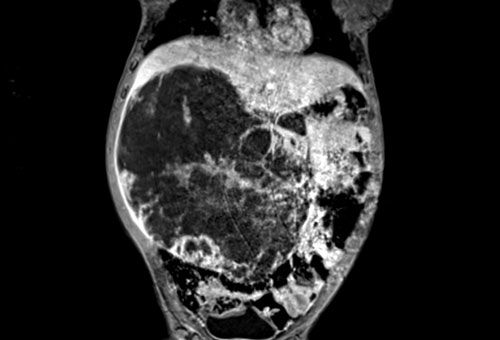
T1 weighted fat suppressed coronal MRI provides a better overview of the liver lesion, which was almost 2 kilograms at resection.
Pathology showed a mesenchymal hamartoma. No further follow-up was necessary.
Example 2
A four-month-old boy presented with an abdominal mass. Ultrasound showed a mass from the caudal part of the right liver lobe, extending into the pelvis.
The mass is not hypervascular.
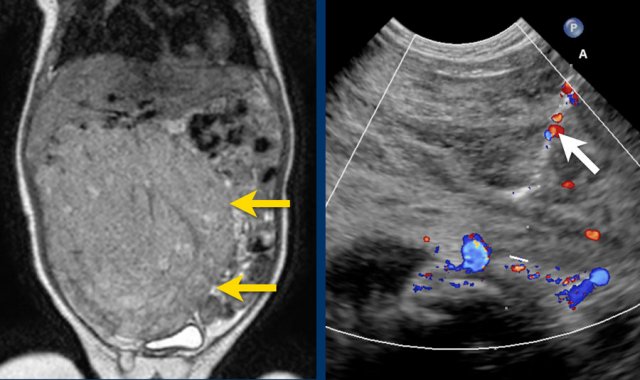
MRI shows a large tumor at the caudal site of the liver, extending to the bladder.
After Gadolinium there was very fast enhancement, almost a arteriovenous shunting.
The mass was thought to be a hemangioendothelioma.
Biopsy was done and pathology was compatible with a mesenchymal hamartoma.
Hepatoblastoma
Hepatoblastoma is the most common malignant liver tumor in young children, while hepatocellular carcinoma presents in older children, mostly in their teens. [7].
Hepatoblastoma usually presents with an enlarged abdomen.
Ultrasound will generally show a well demarcated tumor. In larger tumors necrotic cysts and calcifications can be seen.
CT angiography is done preoperatively to define the relation between the tumor and the hepatic vessels.
MRI will better delineate the tumor. A hepatoblastoma has low signal intenstity on T1 and mixed signal intensity on T2. After Gadolinium patchy enhancement is seen.
Example 1
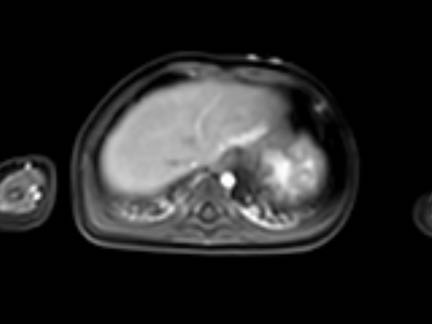
A two-year-old boy presented with a swelling in the abdomen.
On ultrasound a large solid tumor was seen in the upper abdomen. Some calcifications are present. The mass probably originates from the liver. It slides over the right kidney.
Continue with the MRI.
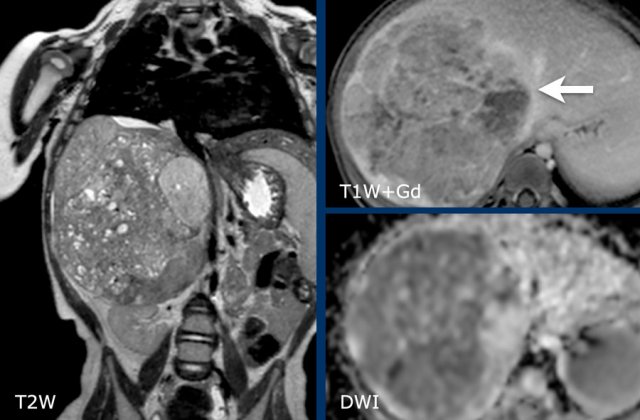
MRI shows a solid hepatic mass with multiple small cysts.
After contrast injection the mass is seen to be limited to the right posterior and anterior section, bordered by the middle hepatic vein (arrow).
The mass has moderate diffusion restriction.
Biopsy was compatible with an epithelial hepatoblastoma.
Hepatocellular carcinoma
This disease is rare in young children but can be seen in older children, mostly >10 years of age, although it has even been reported in children of five years of age. Underlying disease which predispose to HCC include hepatitis B and Tyrosinemia.
Tyrosinemia is a genetic disorder characterized by the failure to break down tyrosine, a building block of most proteins. Tyrosine and its byproducts will build up in organs and can lead to liver and kidney failure and an increased risk for HCC.
The tumor presents with abdominal mass, pain, or jaundice. AFP levels are elevated (although usually less elevated compared to AFP levels in hepatoblastoma).
Example 1
A seventeen-year-old girl presented with upper quadrant abdominal pain. On ultrasound a large liver tumor was detected.
On MRI a tumor in liver segment 5 and 6 is seen with satellite lesions in segment 7 and 8 (arrows). There is a tumor thrombus in the right portal vein (arrowhead), and several lung metastases (*).
Hodgkin and Non-Hodgkin

There are two main types of lymphoma: Hodgkin lymphoma and non-Hodgkin lymphoma.
Hodgkin lymphoma more commonly manifests with cervical lymph node enlargement and mediastinal masses, while it is rarely confined to the abdomen.
Non-Hodgkin is more commonly located in the para-aortic and mesenteric lymph nodes and the spleen (table). Non-Hodgkin lymphoma presents more frequently with extra nodal disease than Hodgkin lymphoma.
For staging of Hodgkin lymphoma the Lugano classification is used [9], and for non-Hodgkin lymphoma the International Pediatric NHL staging system [10].
Ultrasound
On ultrasound enlarged lymph nodes are very hypoechoic. The almost anechoic aspect of the tumor is typical of malignant lymphoma. If the bowel is affected the layering of the bowel wall is lost.
MRI
On MRI masses are seen with some enhancement after Gadolinium and remarkable strong diffusion restriction. Another tumor that can show this marked diffusion restriction is a neuroblastoma, however these tumors are often much more heterogeneous with areas of necrosis and hemorrhage
PET-CT
18-F-FDG PET-CT is used for staging.
Example 1
This video is of a thirteen-year-old boy with cervical and mediastinal locations of a Hodgkin lymphoma.
Sonographic examination of the abdomen demonstrates multiple enlarged hypoechoic paraaortal lymph nodes, which is typical for Hodgkin lymphoma.
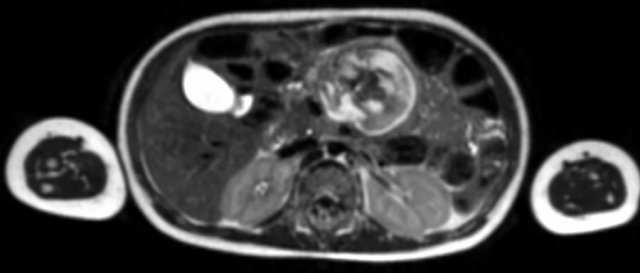
Example 2
A 12-year-old girl presented with a large mass in the abdomen. Ultrasound could not define an organ of origin.
MRI shows a large mesenterial mass and diffuse infiltration of the omentum.

Notice the marked diffusion restriction of the omentum, which makes a lymphoma the most likely diagnosis.
This was confirmed by pathologic examination.
Example 3
A four-year-old suffered from hypertension. His kidneys were enlarged and ultrasound showed a striated appearance of the papilla and hypoechoic areas in the cortex.
There were also enlarged abdominal lymph nodes.
Pathologic examination demonstrated a Burkitt lymphoma.
Leukemia
Leukemia is the most common malignancy in children. It can present with abdominal involvement.
Leukemia can affect all solid abdominal organs.
The organs can be diffusely infiltrated or have a more nodular pattern.
The kidneys are affected in almost half of the patients with later stages of acute lymphoblastic leukemia. It can be uni- or bilateral, and there can be focal lesions or diffuse infiltration. The last has a rather typical appearance with a striated pattern around the calices, like in malignant lymphoma.
Example 1
An eight-year-old girl presented with weight loss and severe pain in the legs.
An ultrasound examination had shown multiple lesions in both kidneys.
MRI demonstrates not only the renal tumors, but also a lesion in the pancreas, right iliac wing, left sacrum and multiple retroperitoneal lymphnodes.
The diagnosis on imaging was malignant lymphoma but the final diagnosis turned out to be leukemia.
Germ cell tumor
The majority of germ cell tumors in children occur in the testis and ovary, but they can arise anywhere in the body, including the abdomen.
They develop from pluripotent stem cells, and therefore have variable cell lines.
Often a mixture of benign and malignant cell lines is found.
The most malignant component on pathologic examination determines the choice of therapy.
The tumor can excrete alpha-fetoprotein and / or beta human chorionic gonadotropin.
Abdominal germ cell tumors are diagnosed because of mass effects.
The most common non-gonadal abdominal germ cell tumor is the sacrococcygeal teratoma.
This entity is discussed on the page Cystic Abdominal Masses in Children.
Germ cell tumors are generally part cystic and part solid. The more solid, the more malignant. [13]
On ultrasound solid tumors are often very inhomogeneous with cystic and solid parts. Calcifications are common.
The mixture of cystic and solid parts suggests germ cell tumor.
On
MRI calcifications are difficult to observe because they have a variable
signal on different MR sequences. They are mostly of low signal intensity and thus hard to notice,
but sometimes they may have high signal on spin echo T1 and T2.
MRI can demonstrate fatty components in the tumor
which strongly suggests the diagnosis of a germ cell tumor.
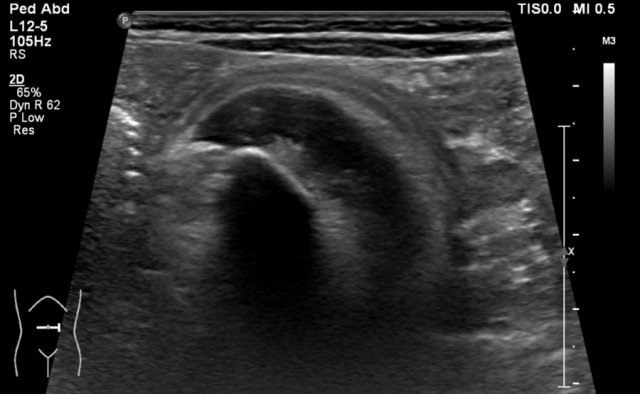
Example 1
A four-month-old boy with an abdominal swelling. Ultrasound shows a tumor with cystic and solid parts. Some echogenic foci are present (arrow).
On MRI (not shown) fatty components were visible.
At operation a teratoma of the stomach was resected. It was benign.
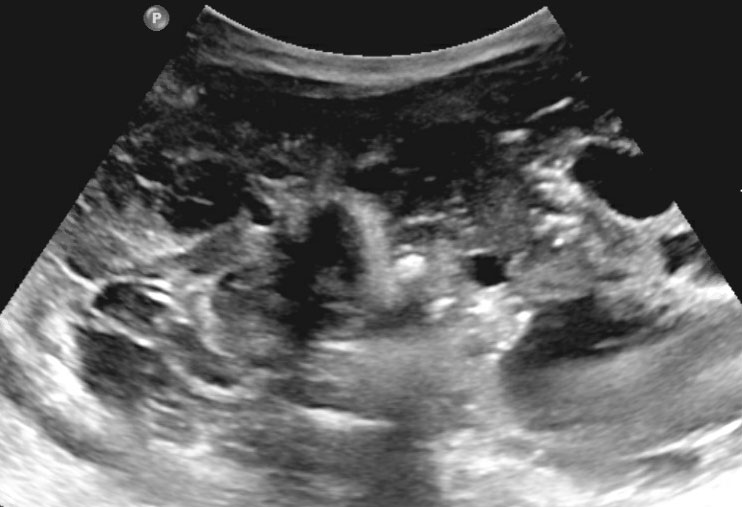
Example 2
A six-month-old boy was suspected of having an intussusception. During the sonographic examination an intraabdominal tumor was seen. He was referred with the diagnosis of a neuroblastoma.
Image
A lesion with a multi-layered wall, a cystic part and an echogenic part with a strong acoustic shadow is present. A germ cell tumor is more likely than a neuroblastoma. Neuroblastoma generally has smaller calcifications.
Continue with the MRI.
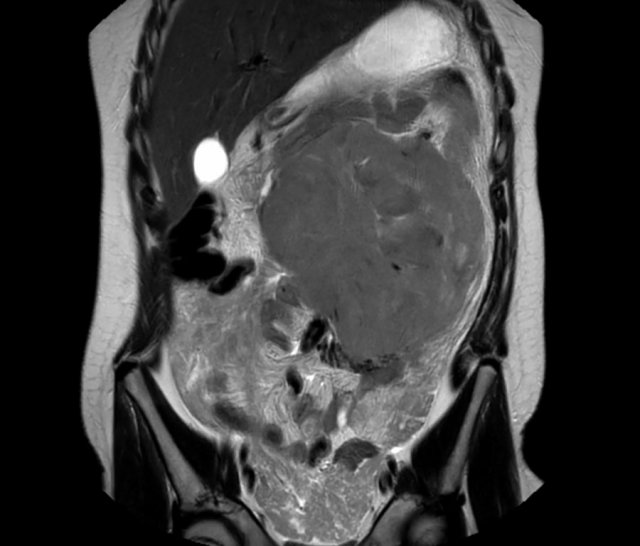
MRI
T2 weighted axial image shows high signal intensity parts of the tumor, either fat or fluid.
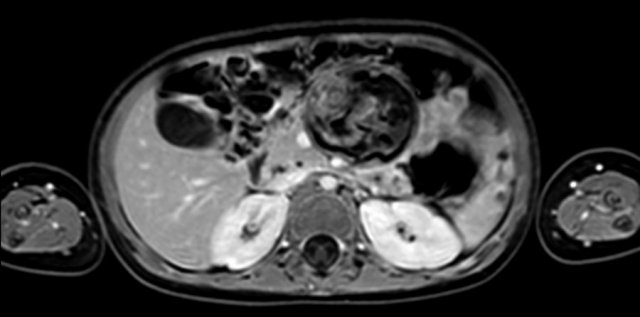
Fat
suppressed contrast enhanced T1W-image.
The
high signal parts are suppressed, which indicates that it is fat.
Note the close relation with the superior mesenteric artery which can be damaged during operation.
At operation a benign mature cystic teratoma was resected.
Example 3
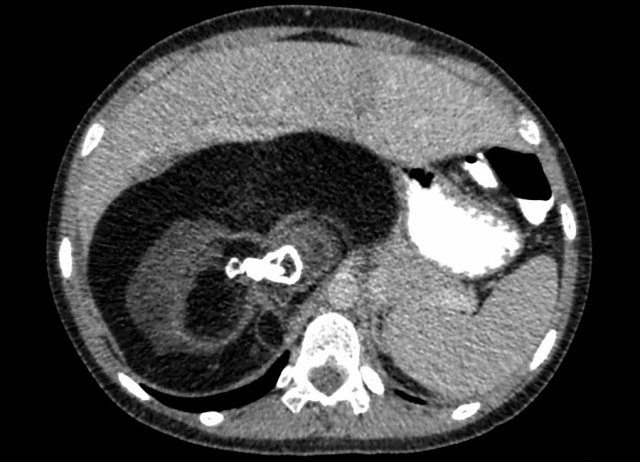
A sixteen-year-old girl presented with swollen legs.
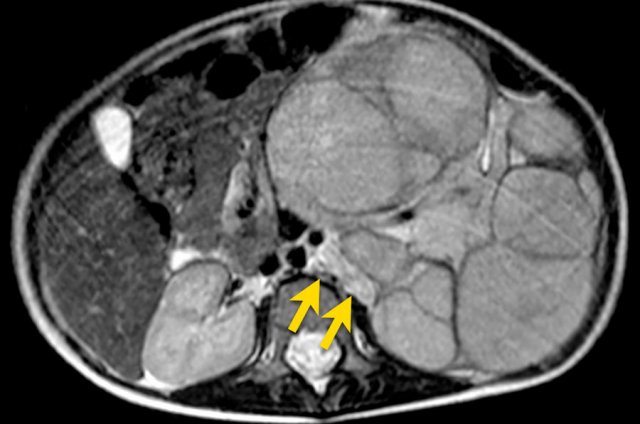
CT demonstrates a mass with fat, coarse calcifications and solid parts.
The inferior caval vein was compressed.
At operation a retroperitoneal mature cystic teratoma was resected.
Rhabdomyosarcoma
Rhabdomyosarcomas (RMS) are the most common soft tissue tumors in children and can develop almost anywhere but mostly in the head and neck region, including the orbit and in the genitourinary tract.
About 25% of all RMS arise in the lower abdomen, generally originating from the bladder, prostate or vagina, but they can arise almost anywhere, for instance along the biliary tract (where no striped muscle is present!).
The most common pathologic subtype is embryonal RMS, followed by alveolar RMS. The alveolar type has a worse prognosis.
The age of the patient, generally below 15 years and the location of the tumor in the prostate, bladder or vagina will point towards the diagnosis, while the imaging features are non-specific.
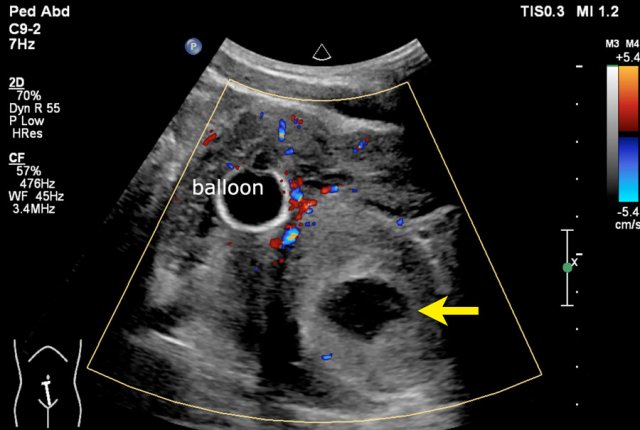
Example 1
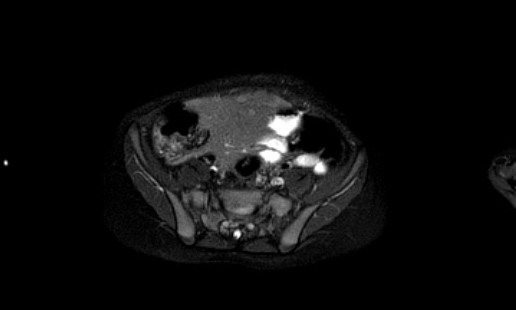
A fifteen-year-old boy with a tumor with a small cystic part (arrow) is seen near the bladder.
Balloon of a catheter in the bladder.
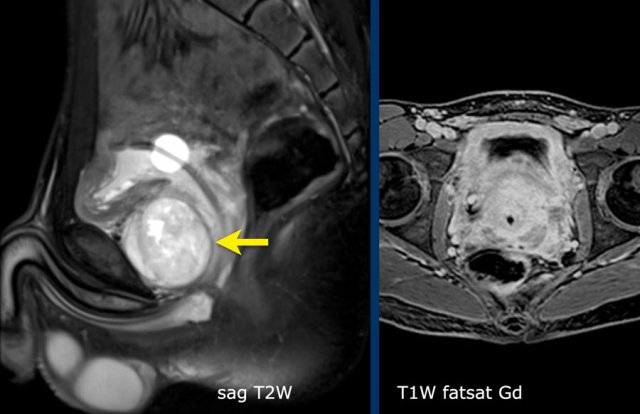
MRI
A sagittal image shows a tumor anterior to the bladder neck.
There
is patchy enhancement.
DWI
showed strong diffusion restriction (not shown).
The location of the tumor makes a rhabdomyosarcoma the most likely diagnosis.
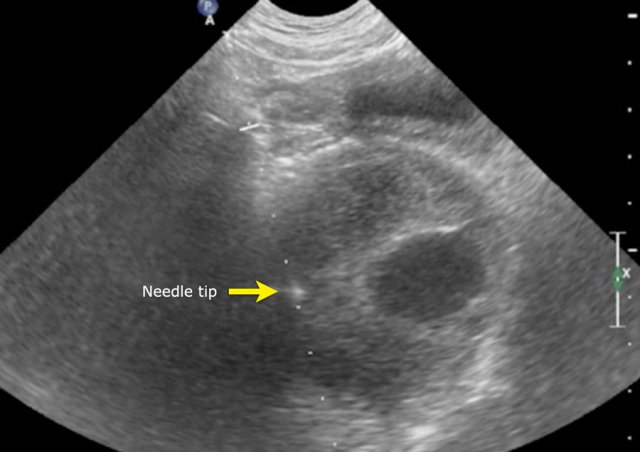
The tumor was biopsied through an anterior approach over the pubic bone.
Final diagnosis: Rhabdomyosarcoma